
[ad_1]
ARYA MASSARAT & MELISSA GYMREK: Describing genetic range with graphs
Reference genomes are essential coordinate techniques for genomic analyses. Nevertheless, the references that scientists presently work from when learning people (the draft human genome1 and its full, gap-free successor2, dubbed T2T-CHM13) are each primarily based totally on single particular person genomes. A linear genome sequence of this kind can’t adequately signify genetic range inside our species. As an alternative, such range is extra precisely described utilizing a graph-based system of branching and merging paths. In a paper in Nature, Liao et al.3 describe the primary human reference pangenome — a group of genome sequences compiled right into a single knowledge construction.
Learn the paper: A draft human pangenome reference
Using human reference genomes from single people is problematic as a result of it introduces biases in how sequences from different human genomes are interpreted. For example, sequences from different genomes are first generally aligned to the reference (learn mapping) after which decreased to a set of variations from that reference (variant calling). Each processes would possibly yield completely different outcomes if a unique individual’s DNA had been used to generate the unique reference. That is notably true for extremely various and structurally advanced areas of the genome. Moreover, there are tons of of megabases of DNA that can’t be captured in a reference primarily based on a single genome, as a result of they exist in solely a subset of people4,5.
A pangenome representing many genomes from completely different ancestries might overcome these points. Nevertheless, developing a pangenome is a posh job. Breakthroughs prior to now decade in long-read sequencing expertise and computational strategies have now enabled this imaginative and prescient to be realized.
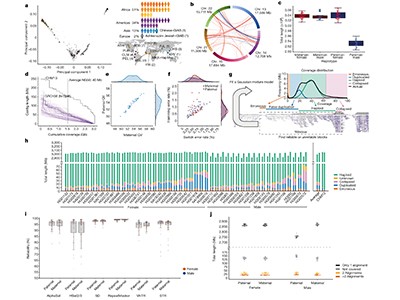
Liao and colleagues first generated 94 genome assemblies from 47 people (one for every of the 2 units of chromosomes that every particular person carries). The people signify various ancestries from across the globe. The assembled genomes, which have been generated utilizing a mix of long-read and different sequencing applied sciences, are extremely correct and practically full, and embrace 119 million base pairs of sequence not included within the draft human reference genome.
The authors used three graph-building strategies to assemble pangenomes from these assemblies. One in every of these strategies aligns all sequences concurrently; the others use one genome as a reference and align every subsequent sequence iteratively. The result’s a set of publicly accessible pangenome graphs, together with a wealthy ecosystem of open-source instruments and standardized file codecs that researchers can use in an identical solution to a linear reference genome.
Liao et al. demonstrated that utilizing their pangenomes for learn mapping and variant calling resulted in 34% fewer errors in calling small variants (these shorter than 50 bases) than did utilizing a linear reference. The distinction was notably pronounced in difficult repetitive DNA areas. Impressively, the pangenomes enabled the authors to establish twice as many giant genomic alterations, referred to as structural variants, per individual than is feasible utilizing a linear reference (Fig. 1).
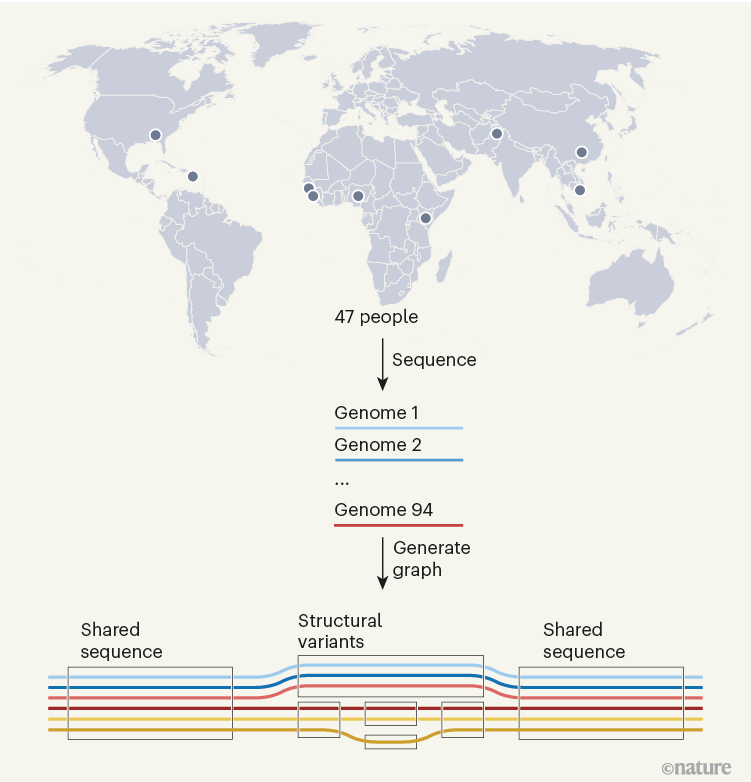
Determine 1 | A human pangenome. The genomes of 47 individuals of various ancestries have been used to generate a draft human pangenome reference3,6,7. Two whole-genome sequences have been generated from every particular person (one for every of their two units of chromosomes). The 94 sequences have been aligned to kind a pangenome graph, which is conceptually much like an underground-railway map. Boxed areas point out sequences current in a number of genomes that at a given web site, with branching paths indicating sequence variation. The graphs reveal giant genome alterations referred to as structural variants, and allow straightforward evaluation of how they range between people.
The human pangenome reference represents a milestone in human genetics. Nevertheless, challenges stay. Alignment of sequences in opposition to extremely variable repetitive areas within the pangenome could possibly be improved by more-accurate assemblies or new algorithms. Extra samples from various teams are additionally wanted. Lastly, widespread adoption of the pangenome by scientists might take time, as a result of new strategies supporting pangenome evaluation are frequently being developed, and scientists will usually require coaching to make use of them.
Continued enhancements in strategies for constructing and utilizing pangenomes will allow researchers to beat these challenges. Use of pangenomes has the potential to rework human genomics. This can finally make it simpler to find genetic variants that mediate bodily and medical traits and — it’s to be hoped — will finally result in higher well being outcomes for many individuals.
BRIAN MCSTAY & HÁKON JÓNSSON: Untangling repeated sequences
Repetitive DNA areas are difficult to sequence, as a result of it’s laborious to put them precisely in a genome meeting. These areas embrace segmental duplications (by which sequences a couple of kilobase lengthy are repeated elsewhere within the genome) and the brief arms (p-arms) of a subset of chromosomes, dubbed acrocentric chromosomes. Two research in Nature now use Liao and colleagues’ pangenomes to systematically discover these areas — Guarracino et al.6 to analyse acrocentric p-arms, and Vollger et al.7 to analyze segmental duplications. Their work supplies a glimpse of the insights that may be gained from a pangenome reference.
Learn the paper: Recombination between heterologous human acrocentric chromosomes
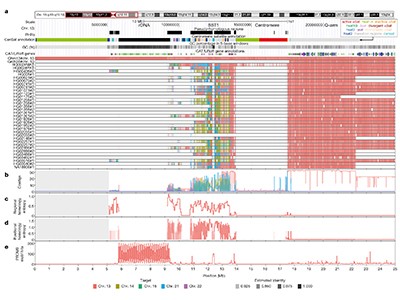
The acrocentric chromosomes (chr13, chr14, chr15, chr21 and chr22 in people) are these by which the p-arm is significantly shorter than the opposite (q) arm. Acrocentric p-arms are devoted to 1 job: forming websites referred to as nucleoli, the place the cell’s protein-assembling machines are made8. P-arms include nucleolar organizer areas (which encode the RNAs that drive nucleolar formation), extremely repetitive DNA and plenty of different shared sequences. This shared, repetitive DNA displays a phenomenon referred to as heterologous recombination, whereby completely different acrocentric p-arms pair and cross over to alternate DNA in the course of the cell divisions that generate sperm and eggs. Against this, in most chromosomes, pairing and crossover are restricted to 2 copies of the identical chromosome (homologous recombination).
In XY intercourse chromosomes, which additionally exhibit heterologous recombination, pairing is aided by brief areas of homology (near-identical sequences) shared between X and Y. Guarracino et al. constructed a variation graph for acrocentric p-arms utilizing Liao and colleagues’ sequences, and located that they include pseudo-homologous areas (PHRs). Every PHR is a patchwork of sequence blocks that — because the authors found once they in contrast their graphs with T2T-CHM13 — usually present extra similarity to the opposite 4 acrocentric p-arms in T2T-CHM13 than to the T2T-CHM13 model of themselves. Presumably, these blocks help heterologous recombination, making certain that p-arms evolve in live performance to protect their shared position in nucleolar formation.
Robertsonian translocations (ROBs) are phenomena, normally occurring throughout egg-cell manufacturing, whereby the q-arms of two acrocentric chromosomes fuse and a lot of the p-arms are misplaced8. Guarracino et al. recognized sequences in PHRs at which the breaks that result in ROBs happen — indicating that ROBs are collateral harm arising from heterologous recombination. On condition that ROBs happen in a single in 800 human births, we surmise that heterologous recombination between acrocentric chromosomes is each ongoing and frequent. We count on that, as extra genomes are added to the pangenome reference, will probably be doable to quantify the frequency of this recombination.
Learn the paper: Elevated mutation and gene conversion inside human segmental duplications
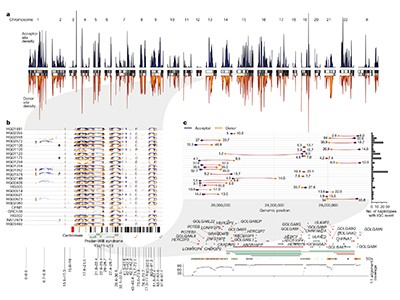
Vollger et al. used the reference to systematically evaluate variation in segmental duplications with that in non-repetitive elements of the genome (Fig. 1). They discovered 60% larger sequence range in segmental duplications, and confirmed that these duplications are extremely divergent between populations and people.
Genes in segmental duplications are vulnerable to interlocus gene conversion (IGC) — an alternate of brief DNA sequences between non-homologous elements of the duplicated area. Vollger and colleagues recognized IGC occasions by in search of indicators of sequence shuffling within the pangenomes, and concluded that these occasions are most likely one of many important causes that segmental duplications are so various. They discovered that 799 genes had protein-coding areas affected by an IGC.
It’s thrilling to see correct characterization of segmental duplications, as a result of duplicated sequences can gas the evolution of recent, specialised roles for a gene. Vollger et al. assessed sequence ‘constraint’ in duplicated genes, with a selected curiosity in these duplicated throughout evolution of the human lineage. Constraint is a measure of sequence variability, with much less variation indicating that mutations are detrimental to the organism’s viability. Thirty-eight genes have been constrained, together with members of the NOTCH2 gene household, which has been linked to human-specific adjustments in mind dimension throughout evolution9. The repetitive nature of segmental duplications had beforehand led to difficulties in assessing constraint for a minimum of 40% of the analysed genes. The authors additionally discovered that 171 genes have been duplicated and relocated intact to new genomic areas, probably which means that their regulation could be rewired. In future, the pangenome undertaking ought to allow researchers to evaluate constraint in not too long ago duplicated genes in additional depth.
Collectively, these papers present a taster of how the human pangenome reference can be utilized. They reveal how sequence exchanges between repetitive areas of our genome contribute to variation within the inhabitants and to our evolution. Because the scope of the reference expands, we sit up for additional insights into these fascinating genomic areas.
[ad_2]
{kind=link}